Tommy Rodriguez
tommy@bioinfoexchange.us
Affiliations: Graduate of University of Maryland UC: Bioinformatics, Graduate School of Environmental Management & Technology, Adelphi, Maryland. Independent Research in the field of Computational Biology. Rodriguez, T. for BioINFO Project, Miami, FL: www.bioinfoproject.com
Keywords: phylogeny, mammalian phylogeny, comparative sequence analysis, mtDNA, multiple sequence alignment, bioinformatics
Abstract
Comparative methods involving DNA-DNA hybridization are thought to be reliable in terms of phylogeny reconstruction, but also weigh heavy on computational resources. Processing large-scale genomic datasets can have potentially unfavorable drawbacks in computing, which can result in poor analysis leading to erroneous conclusions. Slim genomic datasets, such as those found in entire sets of complete mitochondrial DNA (mtDNA), are lucrative options for researchers seeking complete genomic datasets that fit the criteria of a computationally light model. Yet, a handful of incorrect conclusions in molecular phylogenetics have also raised questions about its reliability. This article seeks to emphasize a simplified approach to phylogeny research using complete mitochondrial genomes alone, while touching upon a number of technological perspectives, such as algorithmic selection, which can help improve accuracy and performance in comparative analysis. Moreover, my results will show that reliable estimations can be obtained by using mitochondrial markers, even among time-extended taxonomical rankings; although, admittedly, precise taxonomical boundaries for accurate resolution using complete sequences of mtDNA remain unclear, and it should be explored further. Six distinct mammalian groups of taxa were selected for comparison. In all cases, mtDNA models generated reliable phylogeny approximations when compared against other independent data, while rendering exceptional computational performance.
Computational Considerations
Biological information is compiled of huge amounts of raw data; collecting, processing, and managing biological data can be challenging. Today, modern technology allows advanced next-generation sequencing to be achieved with high resolution and ever-increasing precision. Supercomputers produce optimal results, in minimal timeframes. For purposes of this study, we should work under the assumption of a simplified computer model. That is to say, supercomputers are not required in all cases of phylogeny; and this instance being one of them. In consideration of computing for bioinformatics, the following technological attributes should always be considered: (a) operating system; (b) CPU; (c) physical memory capacity; (d) disk storage; (e) algorithm selection; (f) bioinformatics platform; (g) related software; and (h) networking peripheral(s). Global system adjustments toward bioinformatics usage may be required. This greatly improves the overall experience.
Alas, my models produced a number of results that lean in the direction of fast, efficient computing. Once the mtDNA data was collected and imported (UGENE bioinformatics software), a series of multiple sequence alignment (MSA) tasks were performed on thirteen complete mtDNA sequences of mammalian origin; each sequence represents a unique species within a particular taxonomical group. Here, I selected Kalign for multiple sequence alignment; an accurate and fast MSA algorithm.1 Comparisons done by Lassmann and Sonnhammer (2005) show that Kalign is about 10 times faster than ClustalW and, depending on the alignment size, up to 50 times faster than popular iterative methods.1 Indeed, other comparisons between Kalign and MUSCLE, another popular iterative method, would confirm these results, and show significantly large discrepancies in execution times. The resulting execution time comparisons are reflected in Figure 1 below, where Kalign for MSA yielded regular timeframes of t > 136.15 s and t < 139.95 s, on five separate instances; far superior than MUSCLE, which required exceedingly longer timeframes per interval.
MAFFT is another speedy alternative for MSA. As illustrated in Figure 1, MAFFT would also produce remarkable execution times that are comparable, if not better, to the timeframes produced by Kalign. Yet, in cases of phylogeny involving large-scale genomic datasets with high evolutionary distances, Kalign provides better overall resolution.1 As Lassmann and Sonnhammer (2005) point out, the quality of methods in test sets, namely ClustalW, MUSCLE, and MAFFT, decreased when the number of input sequences was increased.1 This too became evident as I increased the number of taxonomical groups to my working base-pair alignments. In this case, MUSCLE and MAFFT generated a handful of diagrams that were inconsistent with earlier results (containing a reduced volume of sequences). Furthermore, Lassmann and Sonnhammer (2005) concluded the following:
“In order to examine the effects of evolutionary distance and number of sequences, we generated a test set containing 300 alignments. The evolutionary distance was varied in steps of 20 up to 400 and the number of sequences was gradually increased from 20 to 300 sequences. For each individual alignment in this test the winner, i.e. the program with the highest score, was determined. Kalign generally wins in difficult cases of high evolutionary distance and in cases with many sequences … The number of input sequences has a big effect on the running time of each method as the complexity of all alignment algorithms depend on it. Conversely, the more sequences that are used in an alignment, the better an alignment algorithm should perform. To our surprise, the quality of all methods except for Kalign decreased when the number of input sequences was increased. The difference in alignment quality between Kalign and the next best method Muscle reaches 15% at 400 sequences.” (Lassmann and Sonnhammer, 2005) 1
Before proceeding, I should briefly note, the original FASTA data files used in this study did not exceed 220 kb. Light-weight datasets are critical, among other variables that help reduce potential bottlenecks. Now, using our comparative data to examine computational performance, I was able to quantify the amount of useful computational workload compared to the time and resources used.2 Here again, my results would lean toward a Kalign model for MSA. Taken as a whole, Kalign requires the most minimal amounts of resources for execution, in the shortest amount of time.
During runtime, CPU frequency levels peaked at 28.8% and hovered between 26%-28%. At first, the physical memory usage averaged between 25-26 MB, and steadily fluctuated during MSA runtime but did not exceed 26.9 MB. Thus, only 1.2MB of additional RAM was regularly needed to perform MSA on any given instance. In comparison, both MUSCLE and MAFFT far exceeded a computationally efficient mark for physical memory usage, as reflected in Figure 3. Figure 2 also highlights the average range for CPU frequency between the three algorithms, where MUSCLE and MAFFT again exceed the average mark set by Kalign for MSA. Finally, in order to produce a final phylogeny diagram, I ran a UGENE’s tree builder tool on my 17,170 base-pair alignment (Kalign). Here, I implemented the PHYLIP neighbor-joining method coupled together with distance matrix model F84. Regular timeframes for execution would also vary on occasion, and became slightly hindered by bootstrapping compilers.
 |
 |
|
Figure 1 | Execution Time Comparison for Multiple Sequence Alignment: Kalign, MAFFT, and MUSCLE
|
Figure 2 | Average Physical Memory Comparison for Multiple Sequence Alignment: Kalign, MAFFT, and MUSCLE
|
 |
|
|
Figure 3 | Average CPU Frequency Comparison for Multiple Sequence Alignment: Kalign, MAFFT, and MUSCLE
|
|
Measuring Execution Times
Even though measuring execution times is neither straight-forward nor is it achieved with perfect accuracy, each MSA procedure was measured at real-time using the following batch script:
:: Filename: timecmd.bat
:: Written by: Tommy Rodriguez
@echo off
@setlocal
set start=%time%
:: runs command for ugeneui.exe (kalign)
ugene align-kalign --in=FILE PATH --out=FILE PATH
set end=%time%
set options="tokens=1-4 delims=:."
for /f %options% %%a in ("%start%") do set start_h=%%a&set /a start_m=100%%b %% 100&set /a start_s=100%%c %% 100&set /a start_ms=100%%d %% 100
for /f %options% %%a in ("%end%") do set end_h=%%a&set /a end_m=100%%b %% 100&set /a end_s=100%%c %% 100&set /a end_ms=100%%d %% 100
set /a hours=%end_h%-%start_h%
set /a mins=%end_m%-%start_m%
set /a secs=%end_s%-%start_s%
set /a ms=%end_ms%-%start_ms%
if %hours% lss 0 set /a hours = 24%hours%
if %mins% lss 0 set /a hours = %hours% - 1 & set /a mins = 60%mins%
if %secs% lss 0 set /a mins = %mins% - 1 & set /a secs = 60%secs%
if %ms% lss 0 set /a secs = %secs% - 1 & set /a ms = 100%ms%
if 1%ms% lss 100 set ms=0%ms%
:: kalign execution time result
set /a totalsecs = %hours%*3600 + %mins%*60 + %secs%
echo kalign took %hours%:%mins%:%secs%.%ms% (%totalsecs%.%ms%s total)
set start=%time%
:: runs command for ugeneui.exe (MAFFT)
cmd /c ugene align-mafft --in=FILE PATH --out=FILE PATH
set end=%time%
set options="tokens=1-4 delims=:."
for /f %options% %%a in ("%start%") do set start_h=%%a&set /a start_m=100%%b %% 100&set /a start_s=100%%c %% 100&set /a start_ms=100%%d %% 100
for /f %options% %%a in ("%end%") do set end_h=%%a&set /a end_m=100%%b %% 100&set /a end_s=100%%c %% 100&set /a end_ms=100%%d %% 100
set /a hours=%end_h%-%start_h%
set /a mins=%end_m%-%start_m%
set /a secs=%end_s%-%start_s%
set /a ms=%end_ms%-%start_ms%
if %hours% lss 0 set /a hours = 24%hours%
if %mins% lss 0 set /a hours = %hours% - 1 & set /a mins = 60%mins%
if %secs% lss 0 set /a mins = %mins% - 1 & set /a secs = 60%secs%
if %ms% lss 0 set /a secs = %secs% - 1 & set /a ms = 100%ms%
if 1%ms% lss 100 set ms=0%ms%
:: MAFFT execution time results
set /a totalsecs = %hours%*3600 + %mins%*60 + %secs%
echo mafft took %hours%:%mins%:%secs%.%ms% (%totalsecs%.%ms%s total)
set start=%time%
:: runs command for ugeneui.exe (MUSCLE)
cmd /c ugene align --in=FILE PATH --out=FILE PATH
set end=%time%
set options="tokens=1-4 delims=:."
for /f %options% %%a in ("%start%") do set start_h=%%a&set /a start_m=100%%b %% 100&set /a start_s=100%%c %% 100&set /a start_ms=100%%d %% 100
for /f %options% %%a in ("%end%") do set end_h=%%a&set /a end_m=100%%b %% 100&set /a end_s=100%%c %% 100&set /a end_ms=100%%d %% 100
set /a hours=%end_h%-%start_h%
set /a mins=%end_m%-%start_m%
set /a secs=%end_s%-%start_s%
set /a ms=%end_ms%-%start_ms%
if %hours% lss 0 set /a hours = 24%hours%
if %mins% lss 0 set /a hours = %hours% - 1 & set /a mins = 60%mins%
if %secs% lss 0 set /a mins = %mins% - 1 & set /a secs = 60%secs%
if %ms% lss 0 set /a secs = %secs% - 1 & set /a ms = 100%ms%
if 1%ms% lss 100 set ms=0%ms%
:: MUSCLE execution time results
set /a totalsecs = %hours%*3600 + %mins%*60 + %secs%
echo muscle took %hours%:%mins%:%secs%.%ms% (%totalsecs%.%ms%s total)
Using Complete Mitochondrial Genomes in Comparative Analysis of Time-Extended Lineages
For a number of reasons that are well known to molecular phylogenetics, mtDNA is a suitable choice for examining divergence events among closely related species. First, rapid evolution rates in mtDNA produce more molecular variance among immediate populations. This has notable advantages when studying ancestral relationships whose divergence times are thought to be no greater than 8 to 10 Myr. 3 Second, mtDNA is easier to isolate, purify, and sequence than entire sequences of nuclear DNA (nDNA). Each sample cell can contain a thousand copies of mtDNA, and yet only a single copy of nDNA. Lastly, mtDNA degrades slower than nDNA and it contains a higher prevalence in fossilized remains, which allows genetic comparisons of extinct species and closely related non-extinct species.
mtDNA is inherited solely through the maternal line (with a few rare exceptions), and it has an important role in phylogeny research. However, mtDNA comparisons are generally not feasible options for all facets of molecular phylogenetics, especially when reconstructing variation patterns among organisms that span large evolutionary scales. These scenarios will often produce inaccurate results. Consequently, a set of issues arise from using matrilineal lineages to reconstruct evolutionary divergences: (a) rapid rates in base-pair substitution creates saturation that can result in homoplasy; 4 (b) should male and female history differ in a species, then this marker would not reflect the history of the species as a whole but that of the female portion; 5 (c) hybridization can cause mtDNA to move freely between species and may infer incorrect relationships when building phylogenies. 5 6 As pointed out by Coyne (2012) and others, a number of incorrect conclusions due to hybridization effects have raised contention about using mtDNA alone in phylogeny; including a recent paper on the phylogenies of polar bears and brown bears, which resulted in incorrect evolutionary inferences. 5 6 7
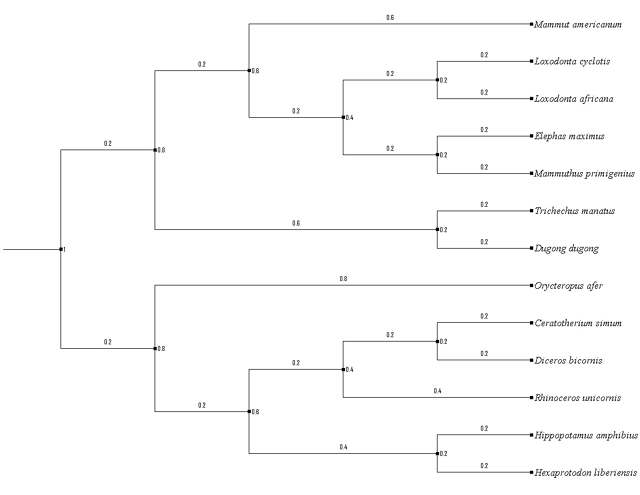
In light of this discussion, it should also be noted: examples of natural hybridization leading to speciation are exceedingly rare, especially in mammals. While most known cases of hybrid speciation occur in plants, the majority of documented cases involving animals have been observed in fish and insects.8 9 Moreover, to some degree, the most effective approach for taking advantage of mtDNA is to combine molecular analysis with other independent data. As it relates to this article, previous studies involving mammalians, and particularly elephantidae and sirenia, have shown congruency in both molecular analysis and comparative morphology. Indeed, my results further confirm this reliable framework, in which divergence events can be assessed by referencing a combination of methods. For one, cases involving comparative morphology of elephantidae strongly support common ancestry between elephas and mammuthus–sister taxa to loxodonata–and extending outward toward mammut americanum. 10 11 12 Molecular analysis combined with comparative morphology (including toenails, molars, prehensile mouth parts, skull, digestive tract, embryology, and gestation periods) would also suggest that manatees and dugongs are among the closest living relatives of the modern elephants. 13 These evolutionary relationships, as described above, were reflected in my phylogenies using comparative mtDNA alone. Please refer to the diagram below.
I expanded upon taxa further to include hippopotamidae, rhinocerotidae, and orycteropodidae, and my results would also match other independent models. Goodman et al. (1981) first identified a set of unique molecular similarities in amino acid sequences of α-crystallin A among aardvark (orycteropodidae ofer), the paenungulates manatee, hyrax, and elephant; and these relationships have since been elaborated further by other sources, namely Honeycutt (2008) and Nishihara et al. (2005). 14 15 16 Orycteropodidae shares a high degree of genetic similarity with three groups: elephantidae, mammutidae, and sirenia, followed by rhinocerotidae; whereas hippopotamida are closely related to modern day cetaceans and contain the largest degree of genetic disparity among the six groups. Morphological data coupled together with fossil evidence would also indicate a series of divergence events between four of the six clades. 17 Once again, the full extent of these evolutionary relationships, as represented from inner node(s) to outer node(s), was well illustrated in the results of my phylogenetic tree and nucleotide distribution patterns. Thus, in some cases, though not all, comparative sequence analysis using complete mtDNA alone can deliver accurate approximates; even in cases involving time-extended lineages.

Figure 4 (above) | Resulting phylogenetic Tree [in no particular order]: (a) Loxodonta cyclotis, (b) Loxodonta africana, (c) Elephas maximus, (d) Mammuthus primigenius, (e) Mammut americanum, (f) Trichechus manatus, (g) Dugong dugon, (h) Orycteropodidae ofer, (i) Ceratotherium simum, (j) Diceros bicornis, (k) Rhinoceros unicornis, (l) Hippopotamus amphibius, (m) Hexaprotodon.
Regarding Sequences and Taxa
Selection of taxa was primarily based on prior knowledge of eutherian mammal phylogeny, while individual sequences were selected based on relative BLAST matches; the BLAST matches were generated via NCBI BLAST tools. I am particularly interested in the evolutionary relationships between elephantidae and sirenia, and their ancestral predecessors; and this also prompted me to include hippopotamidae, due to its semi-aquatic attributes. Each taxa was individually grouped and processed, by way of multiple sequence alignment and hierarchal clustering of phylogenetic trees. The clades were later combined.
Employing the algorithmic configurations previously discussed, I was able to produce a high resolution phylogeny approximation. Reproducibility was initiated on multiple levels; MSA and tree builder operations were repeated several times, in successive order. As mentioned earlier, bootstrapping compilers were applied to hierarchal clustering of phylogenetic trees. During each interval, bootstrap constraints were slightly increased by a ratio of 5% in order to improve my confidence levels in the results; though, each interval yielded nearly identical diagrams after executing a new base-pair alignment using Kalign for MSA. Alas, nucleotide distribution patterns were assembled and analyzed. A series of phylogentic trees details the results of these evolutionary relationships.
Conclusion
Although a handful of incorrect conclusions have raised questions about its reliability, many researchers would still agree that mitochondrial genomes can provide sufficient resolution for reconstructing a robust phylogeny and also facilitate the molecular dating of divergence events within a phylogeny. 18 In terms of approximates, mtDNA becomes particularly useful for species-level and genus-level analysis. 5 Yet, the exact boundary that separates populations among time-extended taxonomical lines based solely on their mtDNA is still not well defined, and should be explored further. And thus, a combinational approach remains the most effective method for taking advantage of mtDNA, where independent data becomes a critical component.
Because computational performance is also tied to a number of instances that have potential bearings on the outcome, I argue in favor of a practical and simplified approach to phylogeny research. Light-weight genomic datasets combined with efficient tools and algorithms for comparative analysis could help reduce potential bottlenecks, make up for lackluster hardware, narrow the scope of error, and enhance our understanding of evolutionary inferences within the spectrum of life. Even though this framework is not at all new to molecular phylogenetics and computational biology, this article reinforces its reliability in both performance and accuracy.
References
1 Lassmann, T., & Sonnhammer, E. L. (2005). Kalign–an accurate and fast multiple sequence alignment algorithm. BMC bioinformatics, 6(1), 298.
2 Allen, A. (1994). Computer Performance Analysis with Mathematica. Academic Press, NY, NY.
3 DeSalle, R., & Giddings, L. V. (1986). Discordance of nuclear and mitochondrial DNA phylogenies in Hawaiian Drosophila. Proceedings of the National Academy of Sciences, 83(18), 6902-6906.
4 Rubinoff, D., & Holland, B. S. (2005). Between two extremes: mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Systematic biology, 54(6), 952-961.
5 Hurst, G. D., & Jiggins, F. M. (2005). Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proceedings of the Royal Society B: Biological Sciences, 272(1572), 1525-1534.
6 Coyne, J. (2012). A new study of polar bears underlines the dangers of reconstructing evolution using mitochondrial DNA. Why Evolution is True. Retrieved from http://whyevolutionistrue.wordpress.com
7 Miller, W., Schuster, S. C., Welch, A. J., Ratan, A., Bedoya-Reina, O. C., Zhao, F., ... & Lindqvist, C. (2012). Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proceedings of the National Academy of Sciences, 109(36), E2382-E2390.
8 Larsen, P. A., Marchán-Rivadeneira, M. R., & Baker, R. J. (2010). Natural hybridization generates mammalian lineage with species characteristics. Proceedings of the National Academy of Sciences, 107(25), 11447-11452.
9 Genner, M. J., & Turner, G. F. (2012). Ancient hybridization and phenotypic novelty within Lake Malawi’s cichlid fish radiation. Molecular Biology and Evolution, 29(1), 195-206.
10 Nishihara, H., Satta, Y., Nikaido, M., Thewissen, J. G. M., Stanhope, M. J., & Okada, N. (2005). A retroposon analysis of Afrotherian phylogeny. Molecular biology and evolution, 22(9), 1823-1833.
11 Orlando, L., Hänni, C., & Douady, C. J. (2007). Mammoth and elephant phylogenetic relationships: Mammut Americanum, the missing outgroup. Evolutionary bioinformatics online, 3, 45.
12 Rohland, N., Reich, D., Mallick, S., Meyer, M., Green, R. E., Georgiadis, & Hofreiter, M. (2010). Genomic DNA sequences from mastodon and woolly mammoth reveal deep speciation of forest and savanna elephants. PLoS biology, 8(12), e1000564.
13 Ripple, J. (1999). Manatees and Dugongs of the World. Voyageur Press.
14 de Jong, W. W., Zweers, A., & Goodman, M. (1981). Relationship of aardvark to elephants, hyraxes and sea cows from α-crystallin sequences.
15 Honeycutt, R. L. (2008). Small changes, big results: evolution of morphological discontinuity in mammals. Journal of biology, 7(3), 9.
16 Nishihara, H., Satta, Y., Nikaido, M., Thewissen, J. G. M., Stanhope, M. J., & Okada, N. (2005). A retroposon analysis of Afrotherian phylogeny. Molecular biology and evolution, 22(9), 1823-1833.
17 University of Calgary (2009, March 19). Is The Hippopotamus The Closest Living Relative To The Whale?. ScienceDaily. Retrieved from http://www.sciencedaily.com /releases/2009/03/090318153803.htm
18 Krause, J., Unger, T., Noçon, A., Malaspinas, A. S., Kolokotronis, S. O., Stiller, M., ... & Hofreiter, M. (2008). Mitochondrial genomes reveal an explosive radiation of extinct and extant bears near the Miocene-Pliocene boundary. BMC Evolutionary Biology, 8(1), 220.